
Strategies for QTL analyses
The aim of QTL analyses is to detect, localize and estimate effects of QTL. The principle of the analyses is to search for non-random associations between phenotypic records and chromosome segments across the genome. Within the segments, the genetic constitution of each animal is deduced from the inheritance of genetic markers. Significant differences in phenotypic expressions between animals with different genetic constitutions indicate the existence of QTL in the studied chromosome segment. For example, consider the simple case of a large half-sib family, whose sire is heterozygous for a QTL and a marker near that QTL (e.g. Q—M and q—m). Offspring inheriting the ‘M’ marker allele (and thus mostly the QTL allele ‘Q’) will have a different mean to those inheriting the ‘m’ marker allele (and thus mostly the QTL allele ‘q’).
In some cases, candidate genes for QTL are known based on information from other populations or other species. Known candidate genes can be tested directly using polymorphisms within the gene or markers closely linked to the gene.
When the aim is to detect unknown QTL, an initial scan of the entire genome has to be performed. In this case markers are genotyped at roughly even spacing across the genome. The genome scan can show the chromosome segments in which QTL are located, but the accuracy of the location is usually low. To increase the precision, and thus improve the possibilities of identifying the QTL, the chromosome segments of interest need to be further studied using other methods, i.e. fine mapping.
|
All phases of QTL mapping (Figure 3) involve analyses of quantitative traits that have a complex genetic background and are influenced by environmental factors. Therefore, in addition to the need for genetic marker information, powerful analyses require good phenotypic records from a large number of animals and the use of suitable quantitative statistical methods. |
|
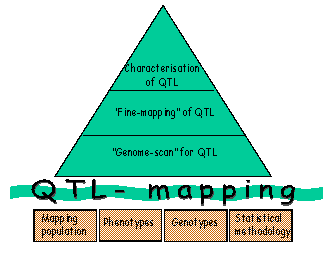
A full genome scan for QTL, aimed at finding the approximate QTL location for subsequent fine mapping and possible use in marker assisted selection (MAS), includes the following steps:
i. Choice of a mapping population: In domestic animals we can either use experimental crosses between divergent populations (such as a breed susceptible to a disease crossed with a breed resistant to the disease) or large families within a population. Studies in designed crosses (e.g. back-cross or intercross designs) are powerful, as they help ensure that family parents (e.g. sires) are heterozygous for the QTL. However, such experiments are expensive for large animals and they do not give any direct answers in relation to the segregation of QTL within the commercial population of interest. The use of families within a population (e.g. a half-sib design) has the advantage that detected QTLs will segregate within the commercial population, but the disadvantage that all sires may not be heterozygous for the QTL.
ii. Collection of phenotype data: To ensure the analysis has sufficient power to detect the QTL(s) of interest, phenotypes are required on a large number of animals. They can either be the same animals that are genotyped or offspring of the genotyped individuals (progeny testing).
iii. Genotyping: Genetic maps, based on DNA markers, are available for many species (see http://www.ncbi.nlm.nih.gov/mapview/). Amongst others, the DNA markers include microsatellites (which are short tandem repeats) and single nucleotide polymorphisms (SNPs; point mutations in the genome). For the genome scan a subset of informative, evenly spaced markers covering the entire genome is selected for the population of interest. The maximum distance between the markers depends on the size of the population and the size of the QTL effects to be detected.
iv. Setting up a genetic model for QTL: Depending on data available, an operational model with one or several QTL (with additive, dominance, epistatic or substitution effects) and remaining genetic and environmental effects is used.
v. Drawing statistical inference from data: The statistical testing for QTL can be performed at marker loci (single marker analysis) or at marker loci and in intervals between markers (interval mapping). In practice, interval mapping is typically used, as in single marker analysis the recombination frequency between the marker and the QTL and the size of the QTL effect are confounded. Different methodologies to test for QTLs include regression, ML and variance component models, amongst others. Due to multiple testing across the genome, permutation testing is typically used to set significance thresholds.
Genome scans usually locate putative QTL to a wide chromosomal region (e.g. 30 to 60 cM). For this reason a genome scan may be followed by a fine-mapping experiment which aims to reduce the confidence region around the QTL to less than a few cMs. Fine mapping typically involves: a) typing more closely spaced markers within the region of interest; b) increasing the experimental population size; and c) use of alternate mapping methodologies (such as approaches based on linkage disequilibrium (LD)). In turn, fine mapping may be followed by experiments aimed at detecting the actual gene of interest and the causative mutation(s) within the gene (see Grisart et al., 2002 as a case study).
Useful references on QTL mapping methodologies include the Armidale Animal Breeding Summer Course notes (2003) and van der Werf et al. (2007).
Genome-wide association analysis
QTL mapping has been successful because of the possibilities to carry out sufficiently large experiments to give a reasonable statistical power for QTL detection. To date, thousands of QTLs have been reported (http://www.animalgenome.org/QTLdb/). However, the identification of the underlying causative mutations remain challenging. Genome-wide association analysis (GWAA) provides a new approach for high resolution genetic analysis, thanks to the development of large panels of SNPs and the development of cost-effective methods for large-scale SNP genotyping and analysis. The number of SNPs required for GWAA depends on the patterns of linkage disequilibrium in the population. Although most domestic animals are not highly inbred, their population structure makes them appropriate for GWAA because they resemble to some extent recombinant inbred lines. Breeds have been developed from large populations by dividing them into many smaller often closed populations on the basis of specific traits. This has led to a reduced genetic diversity within breeds and large haplotype blocks. GWAA surveys most of the genome for causal genetic variants. Because no assumptions are made about the genomic location of the causal variants, the approach could exploit the strength of associations between individual SNPs and phenotypes without having to guess the identity of the causal genes. GWAA therefore presents an unbiased yet fairly comprehensive approach that can be attempted even in the absence of convincing evidence regarding the function of a location of the causal gene. One fundamentally different approach ‘admixture mapping’ could also gain prominence in unravelling the genetic basis of complex traits in domestic animals.
Genomic selection
Recently there has been interest in an approach termed ‘genomic selection’ (GS) as an alternative to the above to identify chromosomal regions of interest for subsequent use in selection decisions. Under GS tens of thousands of SNP markers, closely spaced across the genome such that most or all QTL are in linkage disequilibrium with one marker, are tested for non-random associations with phenotypic records. This is usually performed in a large population—often >1000 individuals representing a large number of families. Useful references for GS methodologies include the Armidale Animal Breeding Summer Course notes (2008), and Goddard and Hayes (2007).
Why map QTL?
The detection and localization of QTL is valuable for several reasons. Firstly, we still know very little about the genetic background of quantitative traits such as growth, muscular development, milk yield, disease resistance etc. Mapping of QTL gives us better insight into the action and interaction of individual genes, which will give us opportunities to refine the genetic models used to describe the variation in quantitative traits. Secondly, associations between genetic markers and QTL can be utilized to improve the efficiency of selection schemes, although this has found limited utility in practice (see Module 3, Section 4.7; Marshall et al., 2009 for a discussion of marker based selection in relation to developing countries). In the case of GS a prediction equation is estimated so that selection of candidates in subsequent generations can be based on genotype information only. Thirdly, mapping of QTL will eventually allow us to identify some of the genes and to study the molecular biology underlying the traits. This knowledge may in the near future be used for genetic modification of genes that are important in breeding programmes, for development of efficient vaccines etc.